by
Ralph C. Merkle
Xerox PARC
3333 Coyote Hill Road
Palo Alto, CA 94304
merkle@xerox.com
http://www.ralphmerkle.com/
Copyright 1995 by Xerox Corporation. All Rights Reserved.
This paper has been published in
Nanotechnology
8, No. 1, March 1997 pages 23-28.
It can be found on the WWW at:
http://www.zyvex.com/nanotech/bindingSites.html.
The web version may differ in some respects from the published
version.
Abstract
Machines, both macroscopic and molecular, are made from parts.
The initial orientation and positioning of those parts is usually random.
To facilitate subsequent parts handling and assembly steps, it is
convenient to position and orient the parts. At the molecular scale,
this corresponds to binding feedstock molecules dissolved in solvent.
In the system design process, the
binding site and the feedstock molecule can be
designed together, simplifying the design of both. Simple
linear molecules (e.g., acetylene, butadiyne, cyanogen) could be
bound by simple tubular binding sites (e.g., bucky tubes).
Planar rectangular molecules could be bound by simple "box"
binding sites. This paper examines several such feedstock-molecule/
binding-site combinations to illustrate that the combined
design process is feasible and often relatively straightforward.
Introduction
Molecular manufacturing should
let us inexpensively fabricate most structures that are consistent
with physical law. One approach for accomplishing this objective
is to design and build self-replicating assemblers, devices
able to precisely position reactive molecular species and construct
complex atomically precise structures by a series of site-specific
molecular reactions. Design proposals for assemblers often include
a barrier which prevents the entry of contaminants and impurities
from the outside environment into the very precise and highly
controlled internal environment. This is the approach taken
in the design of a
"simple" diamondoid assembler (Merkle, 1996),
to name but one example.
If assemblers build things, and the machinery for building things is
protected behind a barrier, this raises the obvious question of how
the raw materials for construction are transported through the
barrier. The purpose of this paper is to begin to
address this question.
Binding site design
The function of a binding site is to (a) bind desired feedstock molecules
from the external solvent, (b) transport those bound molecules into the
interior of the assembler (through a presumably diamondoid wall), (c) block
the entry of undesired molecules and (d) reliably make the bound feedstock
molecule available in a controlled orientation inside the assembler for
further processing.
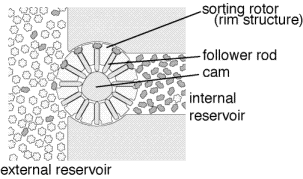 The general proposal in
Nanosystems (Drexler, 1992)
includes multiple stages of filtration
with counter flow. At each stage, desired molecules have a
high probability of moving forwards to the next stage
and a low probability of moving
backwards to the previous stage, while undesired molecules have a high
probability of moving backwards to the previous stage and a low probability
of moving forwards to the next stage. As a consequence, the final stage
will consist almost exclusively of the desired molecules with an impurity
level that can be made arbitrarily low by adding enough stages.
The illustration shows a single selective transporter; many
of these devices would be required for a multi-staged cascade.
The general proposal in
Nanosystems (Drexler, 1992)
includes multiple stages of filtration
with counter flow. At each stage, desired molecules have a
high probability of moving forwards to the next stage
and a low probability of moving
backwards to the previous stage, while undesired molecules have a high
probability of moving backwards to the previous stage and a low probability
of moving forwards to the next stage. As a consequence, the final stage
will consist almost exclusively of the desired molecules with an impurity
level that can be made arbitrarily low by adding enough stages.
The illustration shows a single selective transporter; many
of these devices would be required for a multi-staged cascade.
For the simple diamondoid assembler, we will assume that the cost of the
feedstock can be relatively high (and therefore that the feedstock molecules
themselves can be relatively exotic, if this should prove desirable),
and that the primary objective is to
simplify the design of the binding site and associated transport machinery.
Linear feedstock molecules
Perhaps the simplest binding site would be nothing more than a tube.
For linear feedstock
molecules, such tubes should form good to excellent binding sites.
Bucky tubes form a large class of tubes of varying diameter.
For any arbitrary bucky tube, it would be possible (conceptually)
to cut the tube along its axis and flatten the tube. The resulting
graphitic sheet could then be described by two parameters. Specific
definitions of these two parameters will vary, but possibilities
are the width of the strip (the circumference of the bucky tube)
and its angle with respect to a graphitic sheet; or two indices
giving the end point of the straight line which defines the
circumference (assuming that the starting point of this line
is at (0,0)).
Another way of viewing this is to start with a point on a graphitic
sheet, take an integral number of steps along one crystollographic
axis, followed by another (and typically different) integral number
of steps along the second crystollographic axis, reaching an
endpoint. The straight
line connecting the start point and the end point is then defined
as the circumference
of some bucky tube, while the two integers provide a succinct
name for that bucky tube.
The corresponding bucky tube is obtained
by drawing two parallel lines at right angles to the circumferential
line and passing, respectively, through its start and end points,
and then (a) deleting all atoms not between the two
straight lines and (b) curling up the flat sheet until the two
newly formed edges can be joined.
Clearly, we are most interested in bucky tubes whose circumference is
such that the tube will bind simple linear molecules.
A computer program
to generate the indices for all bucky tubes whose
circumference (or radius) lies
in a given range was written, and a range of tubes surrounding
the estimated best binding radius was generated. The resulting
circumference, radii, index values (which define the
circumference of the bucky tube) and the binding energy
are given.
Circumference (nm) radius (nm) indices Binding energy
to acetylene (maJ, (kcal/mole))
1.9608 0.3121 (8,0) 28 ( 4.06)
2.0062 0.3193 (7,2) 56 ( 8.07)
2.0941 0.3333 (8,1) 96 (13.78)
2.1226 0.3378 (5,5) 118 (16.96)
2.1367 0.3401 (6,4) 118 (16.93)
2.1785 0.3467 (7,3) 120 (17.20)
2.2059 0.3511 (9,0) 125 (18.00)
2.2464 0.3575 (8,2) 123 (17.69)
2.3381 0.3721 (6,5) 115 (16.57)
2.3381 0.3721 (9,1) 121 (17.41)
2.3637 0.3762 (7,4) 114 (16.40)
2.4140 0.3842 (8,3) 119 (15.89)
2.4510 0.3901 (10,0) 108 (15.48)
Table 1. Radii of bucky tubes approximately suited as binding sites
for linear molecules.
The maximally binding bucky tube for this particular potential
energy function is the 9-fold
symmetric bucky tube (an index of (9,0)) as illustrated here:
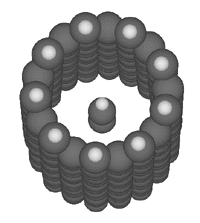
The binding energies shown were computed using the Universal
Force Field (Rappe et al., 1992)
implemented by MSI
in Cerius 2.
The method used was to minimize
a segment of the specified bucky tube, then minimize a segment of
the same bucky tube with acetylene in it. Solvation and entropic
factors were not computed. While these clearly must be considered to
determine the effectiveness of these structures as binding sites
in any particular solvent, the point being illustrated here is not the
specific values but the fact that
the binding energies form a fairly broad basin. This implies that,
even if our current potential energy functions are somewhat in error,
that we can still select a bucky tube which provides a "good" binding
site for a linear molecule.
Some error was likely introduced
because a segment of bucky tube of finite length
and with an irregular hydrogenated termination was used, rather than
an infinite length of bucky tube. Typical bucky tube lengths were on the
order of 20 angstroms, but this varied from tube to tube.
In the (7,3) and more visibly in the larger tubes, the axis of
the acetylene molecule was
misaligned from the axis of the tube after minimization. This is
likely caused by the relatively small van der Waals radius of
the hydrogen atoms at the ends of the acetylene molecule. This
might have contributed to the irregular fluctuations in energy
seen in the larger tubes. Some misalignment appeared to be
present in tubes of smaller radius.
Calculations on C4H2,
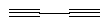 ,
showed that (in accordance with expectations) it
has a significantly higher binding energy than C2H2. For the (8,2)
bucky tube, the binding energy of C4H2 was
210 maJ (30 Kcal/mole),
about 60% larger than the binding
energy of C2H2 for the same bucky tube.
The selective transport of larger molecules should in general be
more reliable (in the sense that the probability that the correct
feedstock molecule has been selected can be made much higher, while
the probability that some incorrect molecule has been selected
can simultaneously be made much lower).
,
showed that (in accordance with expectations) it
has a significantly higher binding energy than C2H2. For the (8,2)
bucky tube, the binding energy of C4H2 was
210 maJ (30 Kcal/mole),
about 60% larger than the binding
energy of C2H2 for the same bucky tube.
The selective transport of larger molecules should in general be
more reliable (in the sense that the probability that the correct
feedstock molecule has been selected can be made much higher, while
the probability that some incorrect molecule has been selected
can simultaneously be made much lower).
 The use of a solvent which is unable to enter the binding site (e.g.,
neopentane)
might also prove useful. In combination with
increased pressure this would
be an effective method of increasing the affinity of the binding
site for the desired feedstock molecule.
The use of a solvent which is unable to enter the binding site (e.g.,
neopentane)
might also prove useful. In combination with
increased pressure this would
be an effective method of increasing the affinity of the binding
site for the desired feedstock molecule.
 (1/9/97)
As an aside,
experimental work by (Thess
et al., 1996) shows that production of (10,10) bucky tubes is
feasible. While the (10,10) bucky tube has a larger diameter than
optimal for linear feedstock molecules, there should be many
bulkier polymers that find it a congenial fit. This suggests,
in particular, that a large "end cap" that could not enter the
(10,10) buckytube could be covalently bound to a polymeric
"tail" that would. An open ended
(10,10) buckytube, when exposed to this hybrid, would
bind to its tail but would leave the end cap
exposed. This might be advantageous in AFM work when
buckytubes are used as nanoprobes,
as there are many possible end caps which (a) could
be covalently linked to a suitable polymeric tail and (b)
would have desirable properties as a probe tip.
(1/9/97)
As an aside,
experimental work by (Thess
et al., 1996) shows that production of (10,10) bucky tubes is
feasible. While the (10,10) bucky tube has a larger diameter than
optimal for linear feedstock molecules, there should be many
bulkier polymers that find it a congenial fit. This suggests,
in particular, that a large "end cap" that could not enter the
(10,10) buckytube could be covalently bound to a polymeric
"tail" that would. An open ended
(10,10) buckytube, when exposed to this hybrid, would
bind to its tail but would leave the end cap
exposed. This might be advantageous in AFM work when
buckytubes are used as nanoprobes,
as there are many possible end caps which (a) could
be covalently linked to a suitable polymeric tail and (b)
would have desirable properties as a probe tip.
Several linear molecules exist. To quote the abstract from
(Lagow et al., 1995):
A carbon allotrope based on "sp" hybridization containing alternating triple
and single bonds (an acetylenic or linear carbon allotrope) has been prepared.
Studies of small (8 to 28 carbon atoms) acetylenic carbon model compounds show that
such species are quite stable (130 degrees to 140 degrees C) provided that
nonreactive terminal groups or end caps (such as tert-butyl or trifluoromethyl)
are present to stabilize these molecules against further reactions. In
the presence of end capping groups, laser-based synthetic techniques
similar to those normally used to generate fullerenes, produced thermally
stable acetylenic carbon species capped with trifluoromethyl or nitrile groups
with chain lengths in excess of 300 carbon atoms. Under these conditions,
only a negligible quantity of fullerenes is produced. Acetylenic carbon
compounds are not particularly moisture or oxygen sensitive but are
moderately light sensitive.
The nitrile terminated
chains are of most interest here, as bulky end caps
would not fit into simple
tubular binding sites of constant diameter. An example of a
short nitrile-terminated linear molecule is cyanogen,
 .
(Safety should also be considered:
cyanogen is highly poisonous,
which is one motivation for our later consideration of
somewhat more complex but less lethal feedstock molecules).
The closing sentence
of the article says "We believe that most
organic end groups and perhaps many inorganic end groups
will stabilize linear carbon."
.
(Safety should also be considered:
cyanogen is highly poisonous,
which is one motivation for our later consideration of
somewhat more complex but less lethal feedstock molecules).
The closing sentence
of the article says "We believe that most
organic end groups and perhaps many inorganic end groups
will stabilize linear carbon."
Another useful property is solubility:
"...capped linear carbon chains are very soluble in most organic
solvents, producing rich amber colored solutions at lower molecular
weights and concentrated dark brown solutions at carbon chain
lengths of about 300." Solvation of feedstock molecules is obviously
important in the system proposal being advanced.
Finally, linear forms of carbon "...should be under appropriate
conditions an excellent precursor for diamond synthesis..."
This is consistent with the observation in Nanosystems
(Drexler, 1992, page 245) that cumulene strands represent promising precursors
for the mechanosynthesis of diamond.
Many short linear compounds involving only first row elements
are well known including:
N2:
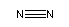
O2:
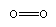
CO2:
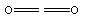
C3O2:
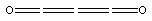
C2H2:
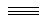
C2N2:
C2HF:
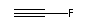
etc. etc.
Other candidates
might include isoelectronic variants of known compounds (e.g.,
CO is isoelectronic with N2).
We can increase the number of linear compounds by including second
row elements, but at the cost of creating a
molecule with a non-uniform radius.
Planar feedstock molecules
 The number of linear feedstock molecules is relatively modest. By contrast,
the number of flat feedstock molecules, though still small in comparison with
the enormous range of possible molecules, represents a much larger class.
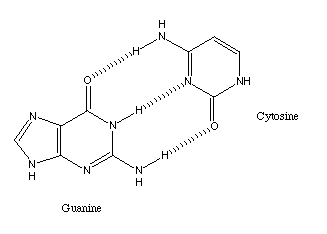
A classic example is the binding between the two DNA bases cytosine and
guanine.
The number of linear feedstock molecules is relatively modest. By contrast,
the number of flat feedstock molecules, though still small in comparison with
the enormous range of possible molecules, represents a much larger class.
A classic example is the binding between the two DNA bases cytosine and
guanine.
While the feedstock molecule is constrained to
be one that can be readily synthesized in bulk, the binding site will be
made using molecular manufacturing techniques, and in particular using
mechanosynthesis. We are therefore not constrained to consider
only binding sites that can be synthesized by present methods, but
are free to consider binding sites whose synthesis would, with present
synthetic methods,
be considered either challenging or actually impossible. As might
be appreciated, this greatly simplifies the task of designing the
binding site.
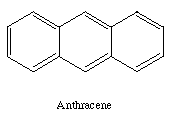 Rectangular feedstock molecules are a particularly simple subset of
the planar feedstock molecules.
As an example, if the selected feedstock molecule is anthracene
a binding site need be little more than a simple box with open
ends into which
the anthracene can slide.
Rectangular feedstock molecules are a particularly simple subset of
the planar feedstock molecules.
As an example, if the selected feedstock molecule is anthracene
a binding site need be little more than a simple box with open
ends into which
the anthracene can slide.
One such box is illustrated here:
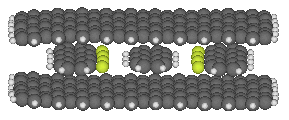
This binding site consists of an upper and lower sheet of
graphite that sandwich the
anthracene between them, and two graphite strips with fluorinated edges
that should have good affinity for the hydrogens along the edge of
the anthracene. All four graphitic components of the binding site
would have to be held in place by further structural elements (not
shown). The placement of the four graphitic components was determined
by minimizing the whole structure.
The binding energy computed with
HyperChem's MM+ potential energy function
(an extension of MM2), and again not considering entropic or
solvent effects, is
285 maJ (41 kcal/mole).
For comparison, the heat of sublimation of anthracene from
25 degrees centigrade is 172 maJ (24.7 kcal/mole) (Dean, 1979).
Anthracene is "slightly soluble" (Lide, 1995) in several
organic solvents (sufficient for our purposes).
It seems likely, therefore, that the binding
site considered here will be energetically preferable to both
the solid and dissolved state of anthracene.
A more complete design for an anthracene binding site is:
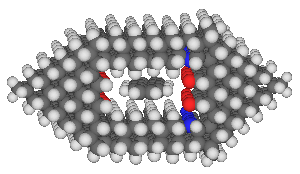
This binding site was designed by positioning two slabs of hexagonal
diamond above and below the anthracene, determining an appropriate
vertical
gap, and then fitting in two end pieces which hold the two primary
hexagonal slabs in the right position. The horizontal gap
was adjusted by selecting among various negative groups
until an acceptable fit was found. Note that the relative
insensitivity of the binding site to minor changes in dimension
(on the order of a few tenths of an angstrom) permitted the design
of a site with a reasonable
binding affinity without requiring extreme precision in the design.
It seems very likely that better binding sites can be designed.
The reader might note that the anthracene is somewhat misaligned
in the site -- this is likely caused by an inability to precisely
adjust the horizontal gap.
This binding site has a predicted binding energy (again using
HyperChem's MM2+ potential energy function) of about 320 maJ
(46 kcal/mole).
As the binding energy is sensitive
both to the particular well depth chosen for the van der Waals energy
and to the particular method of computing electrostatic interactions
(particularly between the F or O and H), it is likely that different
potential energy functions will produce different results. For example,
the Universal Force Field (UFF) (Rappe et al., 1992)
implemented by MSI in Cerius 2, combined
with their algorithm for computing electrostatic interactions,
produces a significantly higher binding energy for both binding
sites. While we can reasonably
conclude that a binding site able to bind anthracene is feasible,
and more broadly that the design of binding sites for a wide range
of other rectangular molecules is likewise feasible, further
research is required to establish the actual binding energy of
any particular binding site with confidence.
The graphitic binding site was minimized
with the anthracene in it, and then minimized again after the anthracene
had been removed. During the second minimization, atoms around the edge
of the binding site were immobilized to prevent the binding site from
collapsing.
It would be theoretically possible for the anthracene to
be caught in a minima after only
partially entering the binding site. For both binding sites,
minimizations started with
the anthracene outside the binding site (but oriented appropriately to
enter the site) resulted in the anthracene entering the binding
site and eventually adopting a central position in the binding site.
While this procedure suggests that any minima other than the principal
minimum are either small or nonexistent, it does not guarantee their
absence.
These binding sites are based both on the general attraction of one molecule
for another (van der Waals forces) and on the principle that opposite
charges attract. By placing
a region of positive charge (hydrogen) in the feedstock molecule close to
a region of negative charge (fluorine, oxygen) in the binding site,
the affinity of
the binding site for the feedstock molecule can be increased.
The major conclusion to be drawn from this example is that binding sites
with relatively high affinity for chosen rectangular
feedstock molecules can be fairly
easily designed given that mechanosynthesis greatly relaxes the synthetic
constraints that would otherwise limit the range of binding site
designs that could be considered. By increasing the size of the feedstock
molecule the binding energy of the binding site can be increased.
If the binding site has a sufficient affinity for the feedstock molecule
it should
be feasible to design a "one pass" binding site and
trans-barrier transport system
that would reliably bind to the chosen feedstock molecule and
reliably reject undesired molecules. While there would be some
probability of error, this probability could be made small
enough that it could be neglected.
There are a reasonably large number of generally rectangular molecules.
There are fluorinated variants of anthracene, such as:
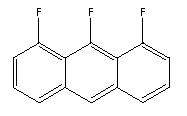
Substitution of N for CH results in molecules like:
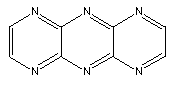
Many more variations are possible.
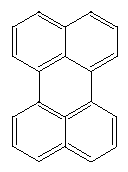
Perylene is another rectangular structure with many possible variations:
The final six membered ring of anthracene could be changed to a five
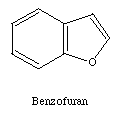
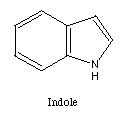
membered ring. Many five membered rings are possible. Two examples
are indole and benzofuran.
The use of five membered rings at the end allows rectangular
molecules which consist of two regions joined in the middle.
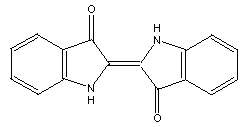
One such structure is indigo:
Another example is the joining of two indoles by a single bond:
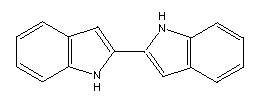
This has the further advantage that structures similar to this
might be used as radical precursors. The two ends of the
structure would be "gripped," mechanical force applied,
and the
relatively weak C-C bond in the middle should break,
yielding two radicals.
Beyond applying force to the two ends
via repulsive overlap interactions (pushing), it is also possible to
bind to appropriately designed molecules with reasonable strength.
One possibility in a vacuum environment would be to use boron
"grippers" that form dipolar bonds to exposed oxygen or
nitrogen (and possibly fluorine).
The dipolar bonds formed between boron on the one hand and
nitrogen or oxygen on the other have the
convenient property that they
are significantly weaker than the covalent bonds that typically
hold a molecule together, but can be much stronger than thermal noise
at room temperature. In a vacuum environment, dipolar bonds
could reversibly bind to small molecules without disrupting those
molecules. This is somewhat reminiscent of
"post-it" notes.
Calculations using the 6-311+G(2d,p) basis set with B3LYP (no zero point
correction) suggest the N -> B bond in HCN -> BH3 is about 19 kcal/mol
(see the Gaussian output for
HCN
-> BH3,
HCN,
and
BH3
for details).
One possible use would be a nitrile group on a diamond (111)
surface with a facing (111) surface in which a boron had been
substituted for a surface C-H. As this would prevent the boron
from assuming a planar configuration, it would tend to increase
the strength of the N -> B dipolar bond.
The generation of radicals (perhaps using the approach suggested
above) would then permit further reactions. For example,
Nanosystems (Drexler, 1992)
proposed the use of an acetylenic radical
to perform selective abstractions of hydrogen, as illustrated
below:
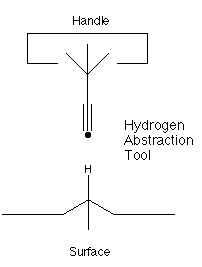
This proposal has been further studied by
(Musgrave et al., 1991) and (Sinnott et al., 1994)
If such a tool is to be used more than once, then the hydrogen must
be removed
from its tip. A proposal in Nanosystems to do this is
to use two weaker radicals (as might be generated by the
mechanism suggested above) to simultaneously attack the C-H bond.
A possible structure for a hydrogen abstraction tool might be
similar to
an anthracene whose end had been modified to resemble the following:
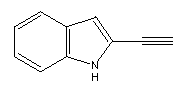
Prior to use, this structure would have to be activated by
removal of the terminal hydrogen.
A more direct method of generating a hydrogen abstraction tool would
be to use a molecule like theone illustrated below:
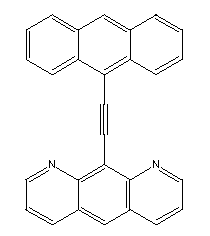
Chem3D Pro's implementation of MM2 predicts that this molecule will
have a slight preference for the planar configuration.
It can reasonably be considered a rectangular molecule and
the design of a relatively high affinity binding site, using the
approach outlined above, should be feasible. The solubility of
this molecule would have to be considered, and variants that had
appropriate solubility might have to be substituted.
Once such a molecule had entered the simple diamondoid assembler,
one anthracene could be rotated 90 degrees with respect to the
other and mechanical force applied, pulling the two anthracenes
apart. While this procedure should rupture one of the
two single bonds, it's not clear exactly which one.
A positionally controlled transition metal might be useful
in preferentially weakening one bond. Other mechanisms
should also be feasible. Without such a mechanism,
it would be necessary to conditionally
select one or the other of the two anthracenes (as appropriate)
in order to get the desired hydrogen abstraction tool.
A variant is 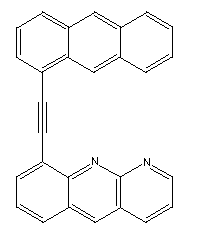
Conclusion
Existing design proposals for diamondoid assemblers (Merkle, submitted
to Nanotechnology) require binding sites able to to bind to
feedstock molecules in solution outside the assembler, and then
move the bound molecule into the assembler for further processing.
The use of planar rectangular feedstock molecules seems to offer a
sufficient range of structures, while at the same time sufficiently
constraining both the search space and the design issues for the
binding sites, that it is worth further investigation.
References
- Dean, J.H. (1979) Lange's Handbook of Chemistry,
Twelfth edition, McGraw-Hill.
- Drexler, K.E. (1992)
Nanosystems: Molecular
Machinery, Manufacturing and Computation, Wiley.
- Lagow, R.J.; Kampa, J.J.; Wei, H.-C.;
Battle, S.L.; Genge, J.W.; Laude, D.A.; Harper, C.J.;
Bau, R.; Stevens, R.C.; Haw, J.F.; and Munson, E. (1995)
Science Vol. 267, January 20, pages 362-367.
Synthesis of linear acetylenic carbon: the "sp" carbon allotrope
- Lide, D.R. (ed.) (1995) Handbook of Chemistry and Physics, 76th
edition, 1995-1996 CRC press.
- Merkle, R.C. (1996) Nanotechnology 7 pages 210-215.
Design considerations for an assembler
- Musgrave, C.B.; Perry, J.K.; Merkle, R.C.; and Goddard, W.A. (1991)
Nanotechnology 2, pages 187-195. Theoretical studies of a hydrogen
abstraction tool for nanotechnology
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.;
and Skiff, W.M. (1992) J. Am. Chem. Soc. 114, 10024-10035.
UFF, a full periodic table force field for molecular
mechanics and molecular dynamics simulations
- Sinnott, S.B.; Colton, R.J.; White C.T.; and Brenner, D.W.
(1994) Surface Science 316, L1055-L1060. Surface patterning
by atomically-controlled chemical forces; molecular dynamics
simulations.
- Thess, Andreas; Roland Lee; Pavel Nikolaev; Hongjie Dai;
Pierre Petit; Jerome Robert; Chunhui Xu; Young Hee Lee;
Seong Gon Kim; Andrew G. Rinzler; Daniel T.
Colbert; Gustavo Scuseria; David Tomanek; John E. Fischer;
Richard E. Smalley; (1996) Science 273,
483-487.
Crystalline Ropes of Metallic Carbon Nanotubes.